What is Optic Atrophy and Cataract type 3?
|
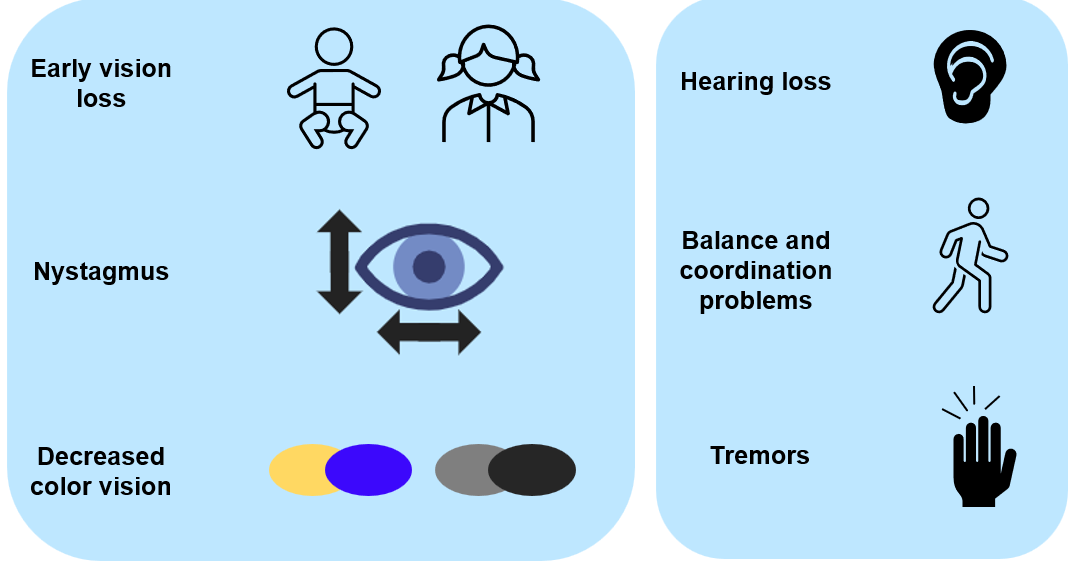
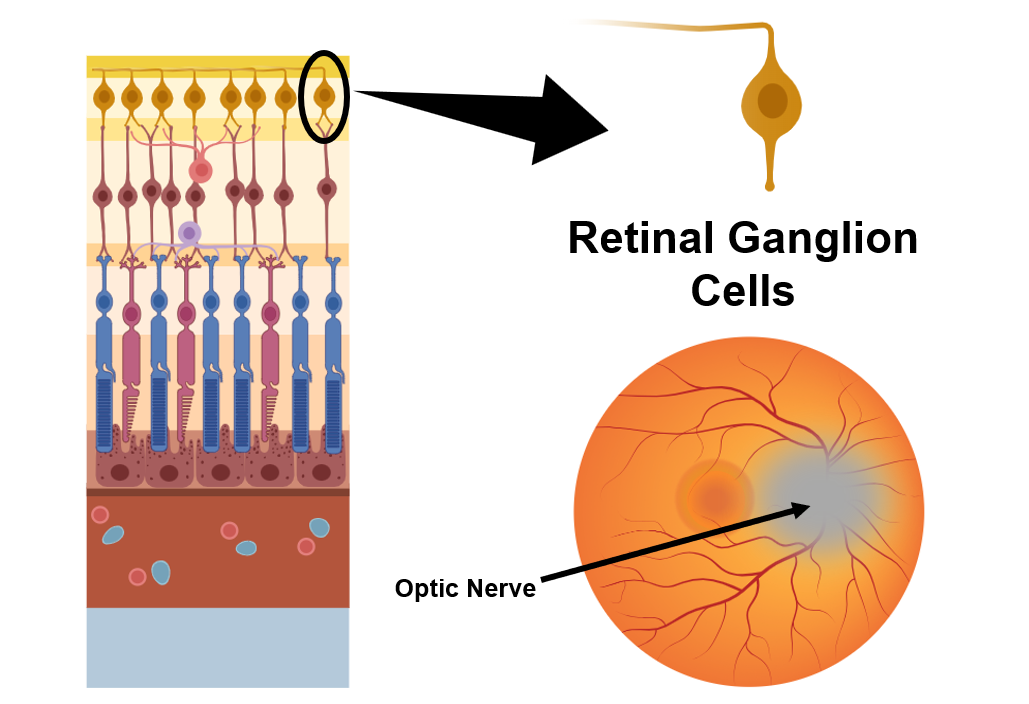
Optic Atrophy type 3 is a genetic disease that is inherited in an autosomal dominant fashion. This means that a carrier of this disease has a 1 in 2 chance of passing on the disease to their child. Optic atrophy type 3 is a rare disease and is a subtype of autosomal dominant optic atrophy (ADOA) that accounts for only ~1% of ADOA cases. The main symptoms are decreased visual acuity from a young age or within the first two decades of life. This is accompanied by nystagmus (uncontrollable, repetitive movement of the eyes) and decreased yellow-blue color vision. Other symptoms can include hearing loss, trouble with gait or coordination and tremors. Currently, there is no treatment available for any ADOA condition. The cells effected in ADOA are within the retina and the optic nerve. Retinal ganglion cells make up the bulk of the optic nerve and carry information from the retina to the brain. These cells atrophy or die causing optic atrophy in the retina which results in the symptoms of ADOA. |
|
OPA3 is mutated in ADOA type 3
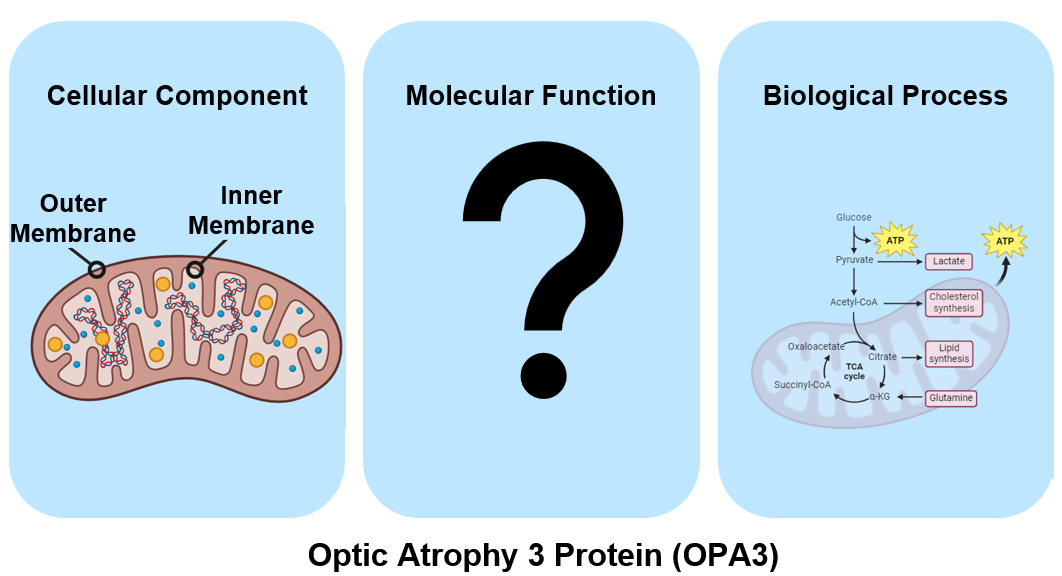
OPA3 or Optic Atrophy Protein 3, is a gene on the long arm of chromosome 19 that has known mutations causing autosomal dominant forms of optic atrophy. The gene ontology for OPA3 is shown below. The protein itself resides in either the inner or outer mitochondrial membrane, different studies conclude different locations. The molecular function has yet to be discovered and is an area of research to be explored. OPA3 is involved in a few different biological processes such as lipid metabolism (shown), apoptosis, and mitochondrial membrane dynamics.
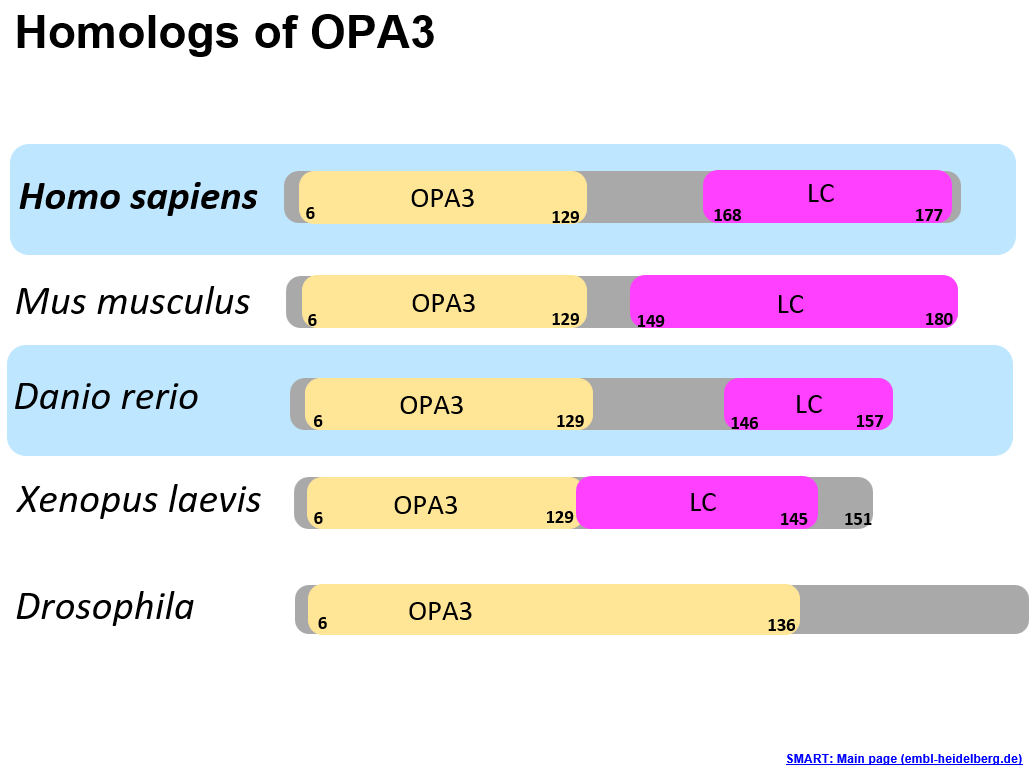
OPA3 is conserved across many species

|
The protein domains within OPA3 are visualized in the image to the left. The yellow OPA3 domain is the most well conserved domain across the model organisms. The pink "LC" domain stands for low complexity and is a domain found by SMART that is predicted to have some molecular function, but the specific function is unknown. This means that the amino acid sequence is important and determining its role could be beneficial in better understanding the disease. Danio rerio, or Zebrafish are an excellent model organism, and their protein domains compare well the human homolog.
The phylogeny tree highlights the relationship between OPA3 between different organisms. As we would expect, OPA3 is very highly conserved in primates but has sequences in many model organisms and Latimeria chalumnae (Coelacanth). Having OPA3 present in many different organisms and an ancient species like Coelacanth suggests some evolutionary importance of the gene. |
OPA3 has few documented protein interactions
|
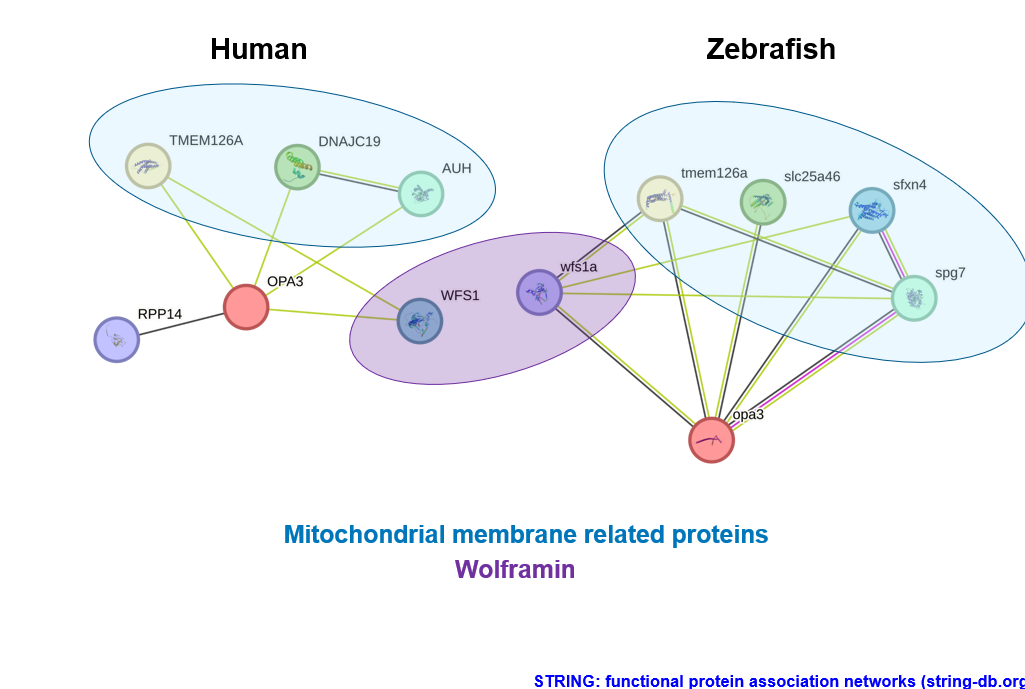
The human and zebrafish protein interactions from STRING are visualized to show what types of proteins OPA3 might be interacting with. In the blue ovals there are mitochondrial membrane related proteins. These are all involved in the structure or maintenance of the mitochondrial membrane and this makes sense give the gene ontology of OPA3. The proteins in purple are the human and zebrafish versions of Wolframin, which is involved in diabetes and has a connection to optic atrophy. Nearly all of these interactions are derived from text-mining (green line) or coexpression (black line). Only one protein interaction is zebrafish was experimentally determined (pink line). More research is needed on protein interactions with OPA3 in optic atrophy type 3 to obtain a basic understanding of the gene's role and to aid in elucidating the molecular function.
|
|
What organism can model Optic Atrophy type 3?
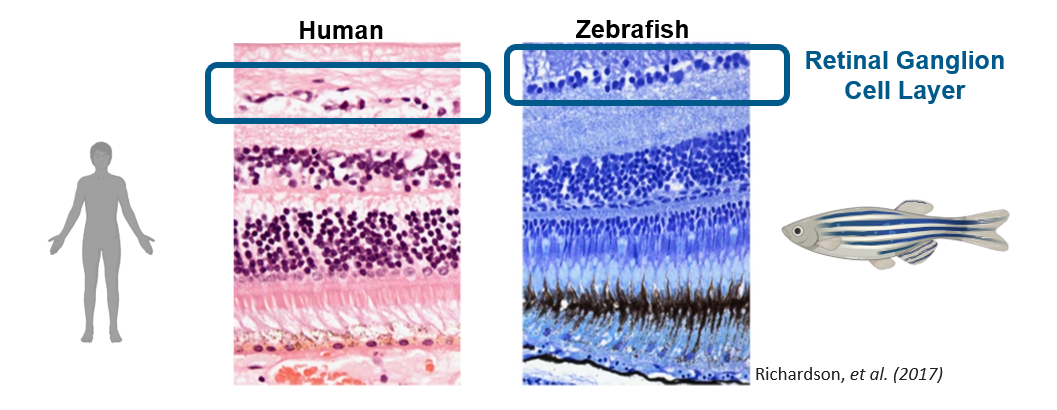
The histological stains above are the retinal cell layers in humans and zebrafish. The morphology is very similar between the two, the retinal ganglion cell layer is the important to the area of research as these cells are mainly affected in optic atrophy. Aside from the physiological similarities, zebrafish are popular organism to study eye diseases and have been used previously in optic atrophy related studies. In addition, zebrafish are relatively cheap, have high fecundity and a short lifespan which all aid in the experimental process.
What is the gap in knowledge?
|
OPA3 is known to have an influence on mitochondrial membrane dynamics and the fission and fusion process. Mutations in OPA3 cause mitochondrial defects such as elongation or fragmentation this is a result of stress to the mitochondria and causes improper energy metabolism in the cell. Currently, there are no studies looking at how mutations in OPA3 are causing these mitochondrial defects in the retina. Furthermore, the connection between mitochondrial defects and retinal ganglion cell health has yet to be explored. |

|
How can we explore the gap in knowledge?
Aim 1: Identify domains in OPA3 that are related to mitochondrial membrane dynamics by performing domain analysis and a CRISPR screen.

Performing domain analysis using programs such as SMART and PFAM allows for the identification of functional sequences in the protein. The OPA3 domain and the low-complexity (LC) domain could have critical functional roles in mitochondrial membrane homeostasis. By understanding the domains and their wild type sequence, it is possible to perform a CRISPR screen for both of the domains to understand what their roles. In doing this experiment we hope to produce OPA3 mutants that exhibit mitochondrial defects and optic atrophy as seen in human patients in the retinal ganglion cells. The hypothesis is that at least one domain, potentially the LC domain, contributes to mitochondrial membrane dynamics in the retinal ganglion cells.
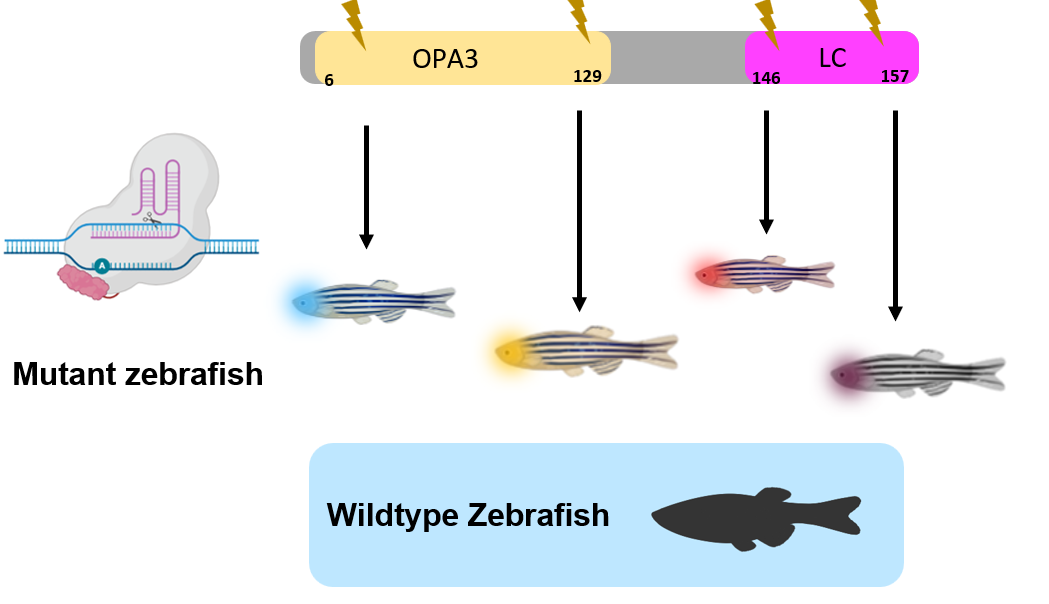
CRISPR screens will be performed in both domains using zebrafish as a model organism. A form of CRISPR that performs base editing can be used to induce single amino acid changes within the domains. Wildtype zebrafish will be used as a control against the mutant fish.
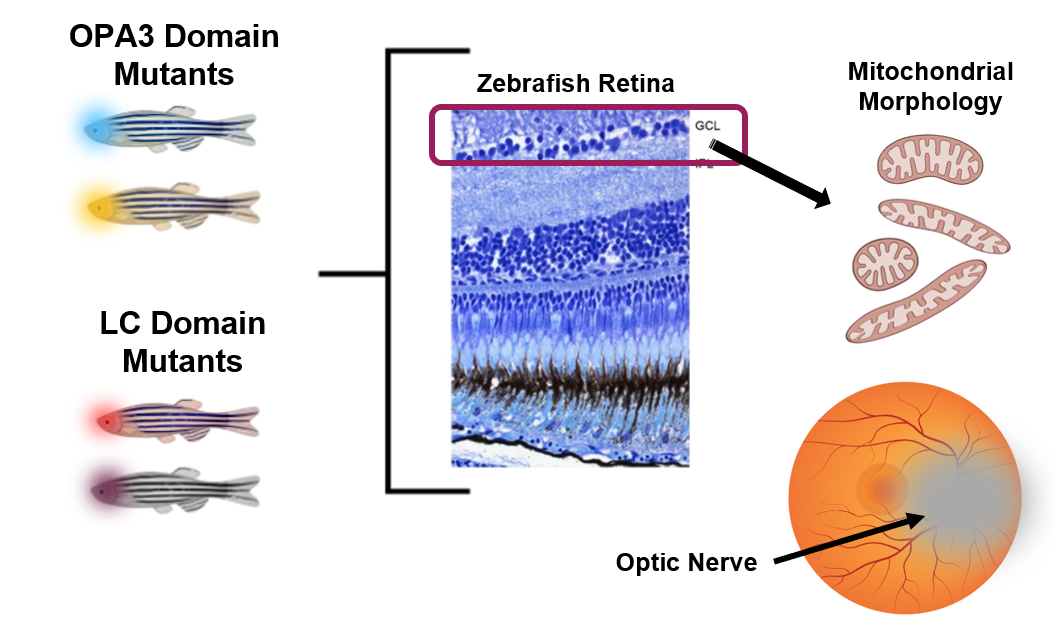
The different zebrafish with mutations either in the LC domain or the OPA3 domain will be phenotypically analyzed to observe differences between the mitochondria morphology and the overall health of the optic nerve. The health of the optic nerve can be analyzed by its visual appearance in the back of the eye and also by use of histological staining, like shown above, to count the number of healthy cells in the retinal ganglion cell layer (GCL). The mitochondrial will be visualized using mitochondrial specific dyes such as mitotracker to analyze defects such as elongation or fragmentation as it pertains to mitochondrial health and membrane dynamics.
Aim 2: Identify small molecules associated with the phenotypic rescue of OPA3 mutants
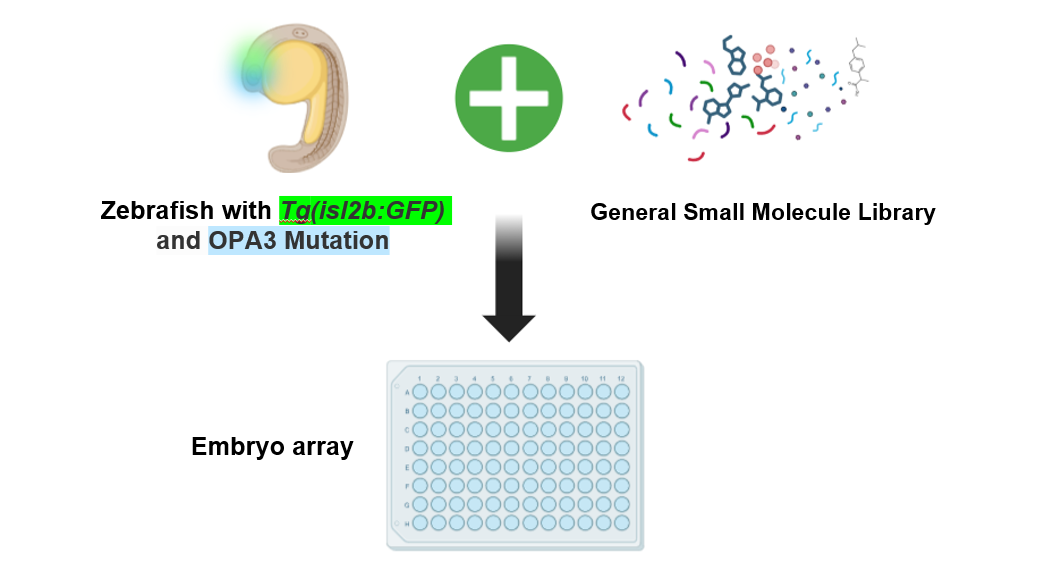
This small molecule screen uses OPA3 mutants, created in aim 1, in conjunction with a general small molecule library carrying different chemicals and drugs. By exposing mutant OPA3 zebrafish to the different small molecules, there is the possibility of reversing or lessening the symptoms of optic atrophy and mitochondrial defects. To assay the health of the optic nerve, transgenic zebrafish with a green fluorescent protein (GFP) only expressed in the optic nerve will be used. Because there are very limited studies on optic atrophy type 3 and no potential treatments, a small molecule treatment could point to potential drugs or mechanistic insights into how the disease works. The hypothesis is that there will be small molecules that inhibit mitochondrial fragmentation and optic atrophy by influencing mitochondrial membrane dynamics.
|
|
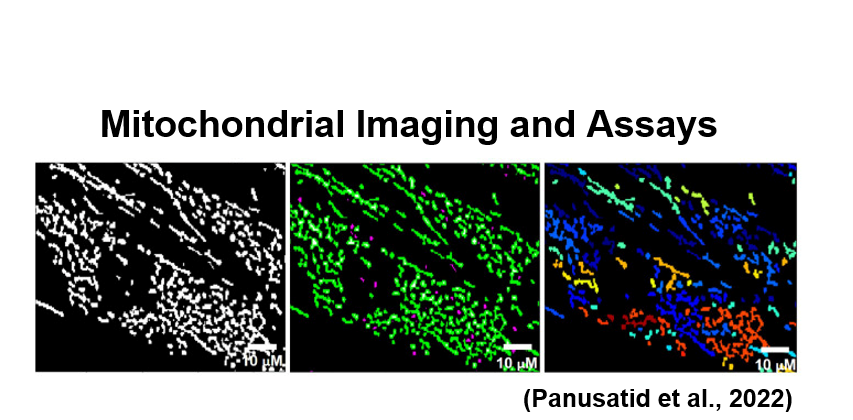
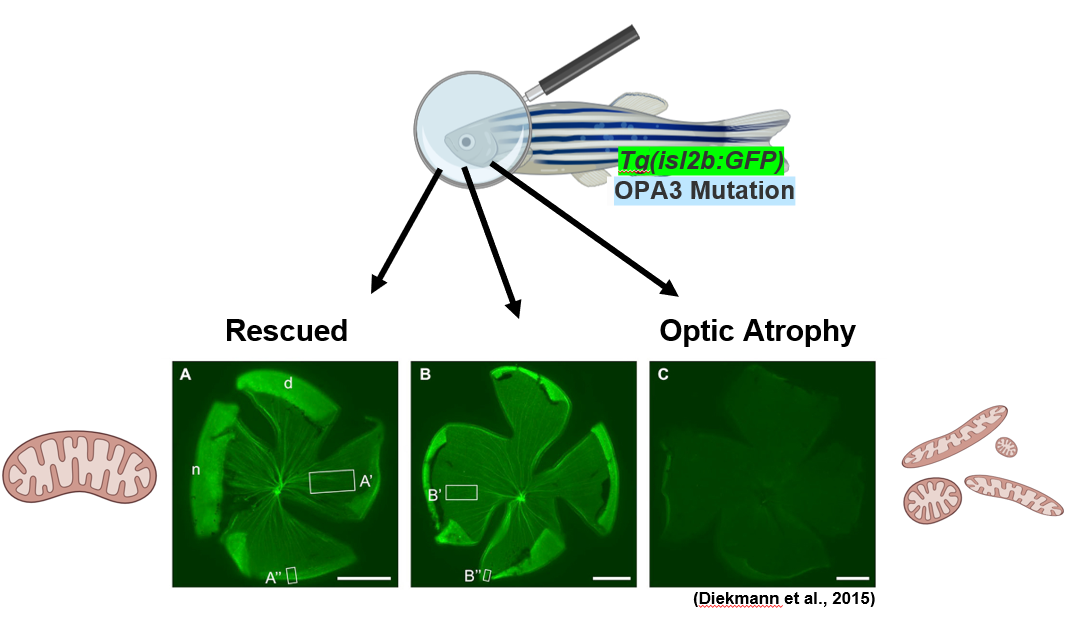
The GFP is expressed in healthy retinal ganglion cells only, which allows for the visualization of the optic nerve in the back of the eye. These retinal flat mounts and their relative fluorescence can be quantified to determine relative optic nerve healthy compared to wildtype zebrafish controls. Mitochondrial imaging within the retinal ganglion cells can also be utilized to determine the mitochondrial shape. In a rescued zebrafish, the optic atrophy and mitochondrial defects would be absent. A healthy or rescued zebrafish retinal flat mount would have bright fluorescence of the retinal ganglion cells like shown in the figure above. An OPA3 mutant that is not rescued would still have the optic atrophy phenotype and appear dull as there is very little GFP being expressed due to the death of retinal ganglion cells. By finding rescued zebrafish and the corresponding drug, it is possible to gain a better understanding about the pathways and molecular functions that OPA3 and optic atrophy type 3 are involved in.
Aim 3: Identify differential protein-protein interactions between wildtype and mutant OPA3 zebrafish
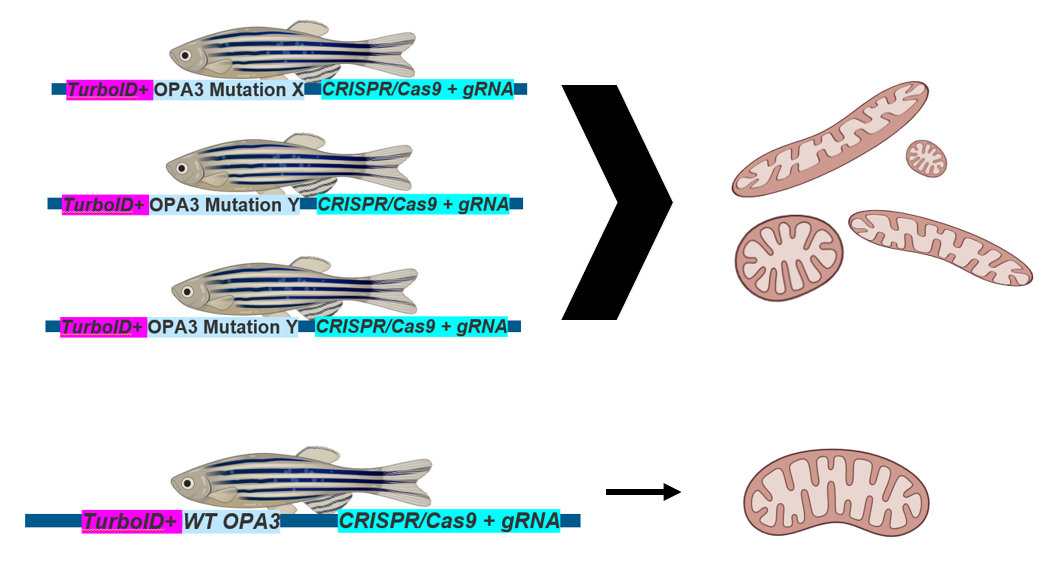
Aim 3 is a proteomic technique that will be utilizing transgenic zebrafish and turboID for proximal protein interaction identification. The using the turboID (pink) is connected to the OPA3 protein sequence of interest, either a mutant form determined in aim 1 or the wildtype form. The turboID tag will allow for increased sensitivity in capturing proximal protein-protein interactions. Between the different mutations in OPA3 mutants and the wiltype zebrafish, we are expecting to see that there are different protein-protein interactions that are causing mitochondrial defects.
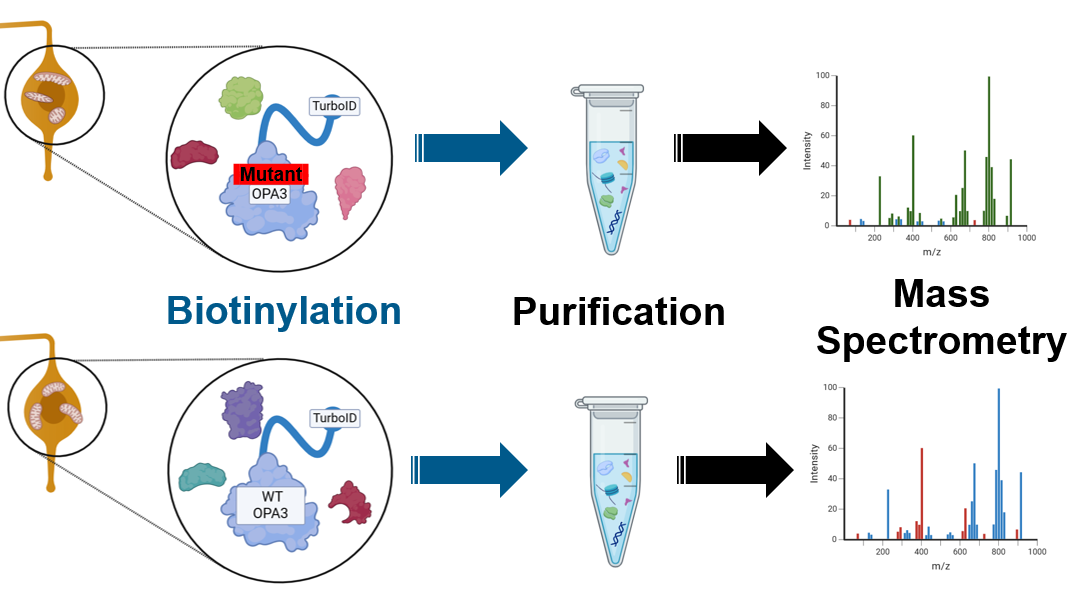
The turboID tag will aid in the biotinylation process which will capture proteins that interact with the OPA3 protein. After purification by streptavidin beads, the proteins from both the mutant lines and the wildtype OPA3 line can be identified by mass spectrometry.
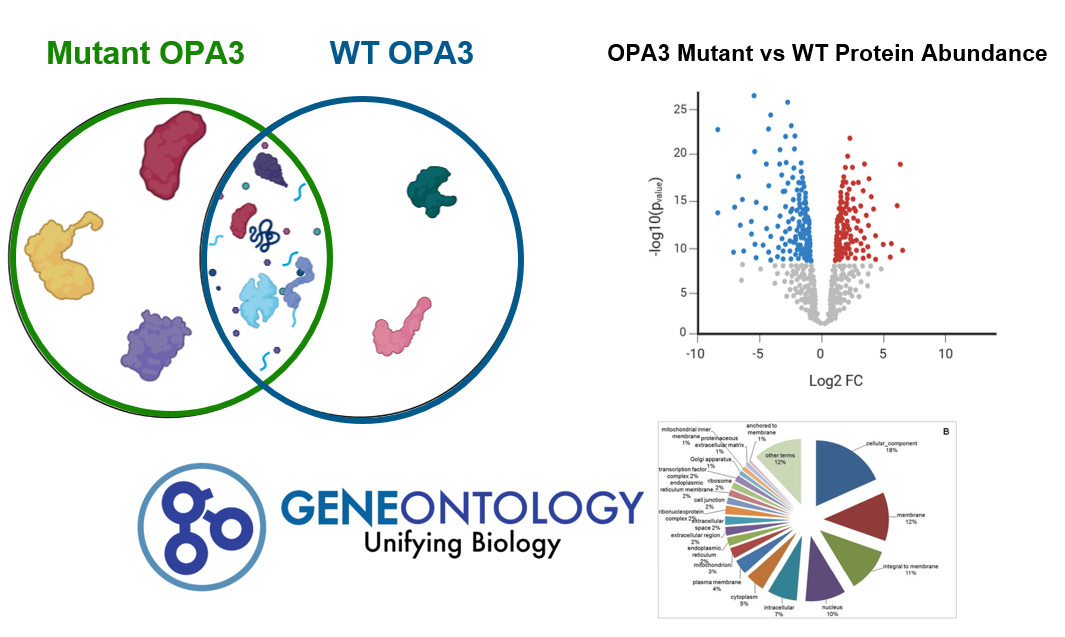
After identifying protein interactions in the wildtype and mutant zebrafish, the results will be sorted and analyzed. The expected result is differential protein interactions between the mutant and wildtype, for example the mutant by have novel or absent protein interactions in comparison to the wildtype (WT). In addition, the abundances of these proteins will be visualized by a volcano plot to analyze how weak or strong protein interactions are between the two types of zebrafish. All proteins will be sorted using gene ontology analysis to identify the pathways OPA3 and its mutant forms are involved in.
Future Directions
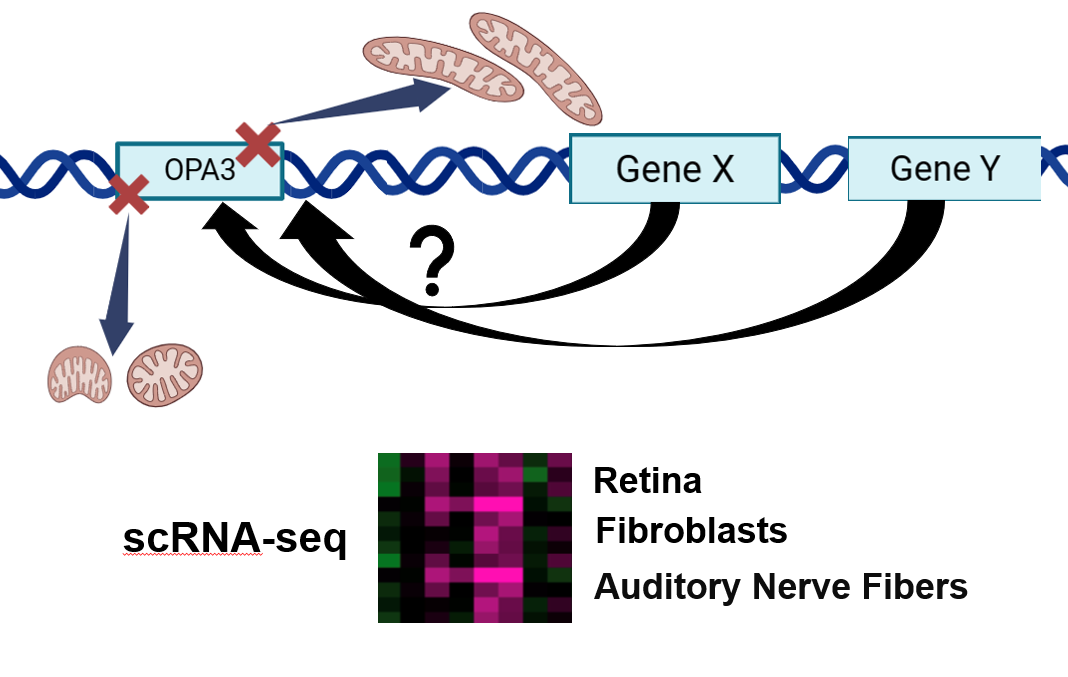
In the future, working towards understanding why OPA3 mutations cause optic atrophy and predominantly affect the eyes and optic nerve could help in revealing more about the disease. While OPA3 is highly expressed in skeletal and muscle tissues, the retina seems to be the most highly affected tissue. Performing scRNA-seq in different tissues could give rise to answers as to why this occurs. Differential gene expression in various tissues likely contributes to mitochondrial defects in the retina. Uncovering the cause and mechanism by which is created could be beneficial in treating this disease as well as other forms of optic atrophy.
|
|
| ||||||
This web page was produced as an assignment for Genetics 564, a capstone course at UW-Madison.
